


Forskerhold udvikler en universel og nøjagtig metode til at beregne, hvordan proteiner interagerer med lægemidler
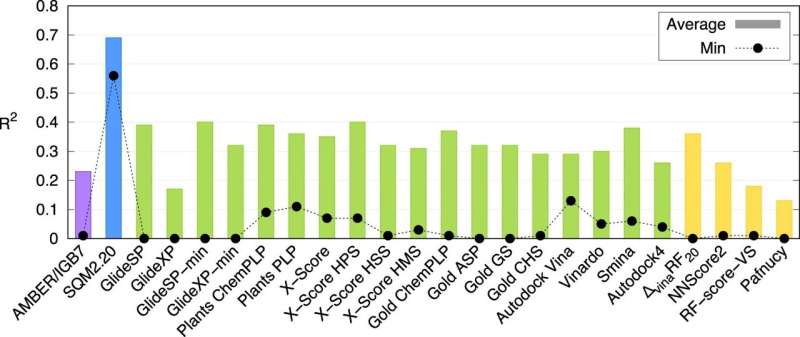
Et forskerhold fra Institut for Organisk Kemi og Biokemi ved Det Tjekkiske Videnskabsakademi / IOCB Prag har udviklet en ny beregningsmetode, der nøjagtigt kan beskrive, hvordan proteiner interagerer med molekyler af potentielle lægemidler og kan gøre det på kun få minutter. Denne nye kvantemekaniske scoringsfunktion kan således markant fremskynde søgningen efter nye lægemidler. Forskningen er blevet publiceret i tidsskriftet Nature Communications .
Undersøgelsen viser, at dette er den første universelt anvendelige metode af sin art. IOCB Prag beregningseksperter testede det på 10 proteiner med forskellige niveauer af strukturel kompleksitet, der hver binder en lang række små molekyler (normalt omtalt som ligander). De sammenlignede derefter deres resultater ikke kun med resultaterne af andre tilsvarende metoder, men også med resultater fra laboratorieforsøg, og begge sammenligninger viste sig meget positivt.
"Selvfølgelig er vi ikke de eneste, der arbejder på dette. Der er flere sådanne metoder. Normalt bliver deres hastighed dog opvejet af lav nøjagtighed, hvorimod mere nøjagtige beregninger kan tage flere dage. Vores metoder er unikke ved, at de kan behandle information om store molekylære systemer inden for ti minutter og samtidig bevare fordelene ved meget mere krævende kvantemekaniske beregninger," forklarer Jan Řezáč, tilsvarende forfatter til artiklen fra gruppen Non-Covalent Interactions ledet af prof. Pavel Hobza.
Eksperter fra denne gruppe har studeret intermolekylære interaktioner i lang tid. I denne forskning fokuserer de hovedsageligt på biomolekyler, og resultaterne af deres arbejde har direkte indflydelse på computerstøttet design af lægemidler. Årsagen er, at når forskere arbejder hen imod et nyt lægemiddel, leder de ofte efter molekyler, der binder stærkt til et bestemt protein.
At identificere dem er imidlertid beslægtet med at finde nåle i en høstak, da et stort antal molekyler skal testes for at adskille dem, der viser lovende. Dette bremser opdagelsen af medicinske stoffer betydeligt og gør det dyrere. Ved at forudsige styrken af protein-ligand-binding og dermed udskille molekyler, der bedst opfylder et defineret sæt kriterier, skåner beregningskemikere forsøgsledernes arbejde, hvilket igen fremskynder opdagelsen af lægemidler betydeligt.
Flere oplysninger: Adam Pecina et al, SQM2.20:Semiempirisk kvantemekanisk scoringsfunktion giver DFT-kvalitet protein-ligand-bindingsaffinitetsforudsigelser på få minutter, Nature Communications (2024). DOI:10.1038/s41467-024-45431-8
Journaloplysninger: Nature Communications
Leveret af Institute of Organic Chemistry and Biochemistry i CAS
 Varme artikler
Varme artikler
-
 Forskere syntetiserer analoger af stoffer, der bruges i lægemidlerRøntgendiffraktionsanalyse af en af forbindelserne beskrevet i artiklen. Kredit:Tatiana Borisova RUDN University kemikere har syntetiseret nye isoquinolinderivater. På grund af deres biologiske
Forskere syntetiserer analoger af stoffer, der bruges i lægemidlerRøntgendiffraktionsanalyse af en af forbindelserne beskrevet i artiklen. Kredit:Tatiana Borisova RUDN University kemikere har syntetiseret nye isoquinolinderivater. På grund af deres biologiske -
 Sukkermetabolisme er overraskende konventionelt ved kræftGrafisk abstrakt. Kredit:Molecular Cell (2022). DOI:10.1016/j.molcel.2022.07.007 I over et århundrede har kræftcellernes stofskifte været betragtet som noget af et paradoks. Nyt arbejde fra forsker
Sukkermetabolisme er overraskende konventionelt ved kræftGrafisk abstrakt. Kredit:Molecular Cell (2022). DOI:10.1016/j.molcel.2022.07.007 I over et århundrede har kræftcellernes stofskifte været betragtet som noget af et paradoks. Nyt arbejde fra forsker -
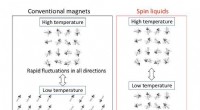 Fra rigelige kulbrinter til sjældne spinvæskerFig. 1 Forskelle mellem almindelige magneter og centrifugeringsvæsker. Ved høje temperaturer, spinnene - de små kompasser for hver uparret elektron i materialerne - svinger tilfældigt mellem vilkårlig
Fra rigelige kulbrinter til sjældne spinvæskerFig. 1 Forskelle mellem almindelige magneter og centrifugeringsvæsker. Ved høje temperaturer, spinnene - de små kompasser for hver uparret elektron i materialerne - svinger tilfældigt mellem vilkårlig -
 Korrekt passende brandhæmmende arbejdstøj til kvinder(Fra venstre) Tekstilforskere Patricia Dolez, Ankita Shroff og Mahsa Kalantari arbejder sammen med iværksætteren Jess Black for at skabe et slidstærkt brandhæmmende stof, der kan bruges i kvinders arb
Korrekt passende brandhæmmende arbejdstøj til kvinder(Fra venstre) Tekstilforskere Patricia Dolez, Ankita Shroff og Mahsa Kalantari arbejder sammen med iværksætteren Jess Black for at skabe et slidstærkt brandhæmmende stof, der kan bruges i kvinders arb
- Overtone
- Video:Det tusind år gamle æg
- Hvorfor videnskabsmænd er fascineret af luft i NASAs Mars-prøverør
- På jagt efter gigantiske planetanaloger i vores egen baghave
- Protestware er i fremmarch, hvor programmører selvsaboterer deres egen kode. Skal vi være bekymred…
- Otago-videnskabsmand udgraver middelalderlig usbekisk kirkegård