


Brug af AI til at identificere genomiske afvejninger mellem typer af mutationer
Grafisk abstrakt. Kredit:Cell Reports (2022). DOI:10.1016/j.celrep.2022.111351
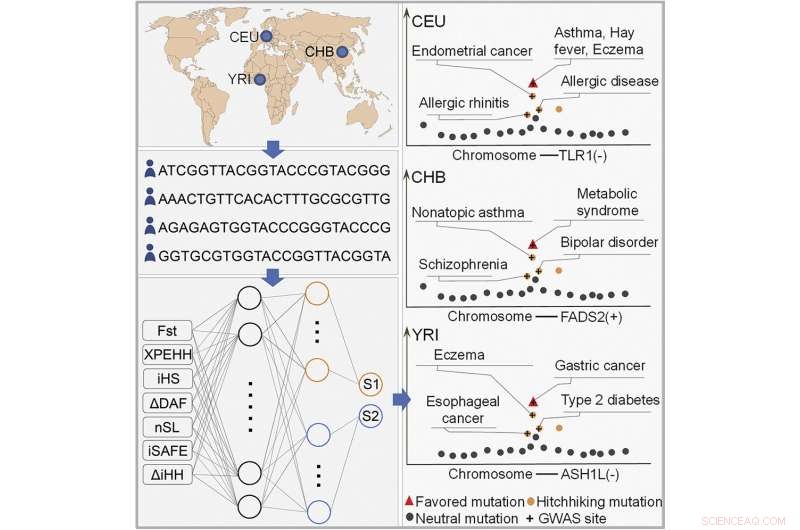
Et team af forskere ved Southern Medical University har udviklet en AI-applikation til at hjælpe med at identificere genomiske afvejninger mellem forskellige typer mutationer, der opstod efterhånden som mennesker udviklede sig. I deres papir offentliggjort i tidsskriftet Cell Reports , beskriver gruppen, hvordan de brugte data fra nuværende eksisterende genom-dækkende associationer til at lære deres system, og hvad det viste, når de blev udsat for nye data.
Tidligere forskning har vist, at efterhånden som væsner udvikler sig, opstår mutationer. Mutationer, der forbliver i genomet, er det, der får et væsen til at udvikle sig. Tidligere forskning har også vist, at nogle mutationer fører til direkte fordele, såsom evnen til at behandle visse fødevarer, hvilket tillader et væsen at eksistere i et nyt miljø. Andre mutationer, på den anden side, tager nogle gange bare med på turen. De giver ikke nødvendigvis nogen fordele, men forbliver i genomet ved et uheld eller på grund af deres nærhed til gener, der giver en fordel.
Forskere, der studerer genomet, har længe ønsket et værktøj, der kunne bruges til at bestemme, hvilke mutationer i det menneskelige genom, der var favoriseret, og hvilke der blot var blaffere. I denne nye indsats har forskerne udviklet et sådant værktøj, selvom det stadig ikke er klart, hvor nyttigt det virkelig er.
Kaldet DeepFavored, værktøjet blev skabt ved at udvikle et dybt indlærings-AI-system, der blev fodret med data fra eksisterende genom-dækkende associationsstudier for at lære af erfaringerne fra andre forskere, der arbejder på tidligere specifikke indsatser. De, der var inkluderet af holdet, blev indsnævret til alleler relateret til kost og andre metaboliske aktiviteter, og også mutationer, der gjorde det muligt at håndtere variationer i klimaet – fokus var på menneskers unikke evne til at tilpasse sig så mange forskellige dele af planeten. Holdet kørte derefter værktøjet på tre separate populationer og fandt, hvad de beskriver som eksempler på blaffemutationer, der har ført til sygdomsmodtagelighed.
Ved at teste deres nye værktøj fandt forskerne også, hvad de beskriver som foretrukne mutationer blandt de tre populationer, de testede. De foreslår, at deres overordnede resultater indikerer, at deres værktøj er i stand til at finde beviser for mutationelle afvejninger i det menneskelige genom. De sammenlignede også resultater fra DeepFavored med to andre algoritmer skabt af andre teams og fandt ud af, at de klarede sig bedre end begge. + Udforsk yderligere
Brug af maskinlæring til at finde mutationer i lignende genomsekvenser af cancerprøver
© 2022 Science X Network
 Varme artikler
Varme artikler
-
 Hvad er røgelse og myrra?I Bibelen, de tre vise mænd bringer gaver af guld, røgelse og myrra til Jesusbarnet. Men hvorfor de to krydderier? Kevin Smart/Getty Images Hvis du har hørt om røgelse og myrra, Det er sandsynligvis
Hvad er røgelse og myrra?I Bibelen, de tre vise mænd bringer gaver af guld, røgelse og myrra til Jesusbarnet. Men hvorfor de to krydderier? Kevin Smart/Getty Images Hvis du har hørt om røgelse og myrra, Det er sandsynligvis -
 Forhistoriske kvindeskeletter viser virkningen af strengt manuelt arbejdeKredit:Shutterstock Kvinder, der levede for omkring 7.000 år siden, gjorde en masse tunge løft i deres tidlige agrariske samfund. Nu afslører skeletanalyse, at de var endnu stærkere end de bedste
Forhistoriske kvindeskeletter viser virkningen af strengt manuelt arbejdeKredit:Shutterstock Kvinder, der levede for omkring 7.000 år siden, gjorde en masse tunge løft i deres tidlige agrariske samfund. Nu afslører skeletanalyse, at de var endnu stærkere end de bedste -
 Twin Monkeys første gang nogensinde klonede som Dolly fåretTo klonede makakker ved navn Zhong Zhong og Hua Hua holdes af en sygeplejerske på det kinesiske videnskabsakademi den 22. januar, 2018. Tvillingeprimaterne er verdens første til med succes at blive kl
Twin Monkeys første gang nogensinde klonede som Dolly fåretTo klonede makakker ved navn Zhong Zhong og Hua Hua holdes af en sygeplejerske på det kinesiske videnskabsakademi den 22. januar, 2018. Tvillingeprimaterne er verdens første til med succes at blive kl -
 Støjforurening forårsager kronisk stress hos fugle, med sundhedsmæssige konsekvenser for ungeDenne vestlige blåfugl vogter sin klø af æg i en redekasse i et naturgasfelt. Ude af stand til at skelne, om deres miljø er sikkert på grund af kompressorstøj, fuglemor skal vælge mellem at holde sig
Støjforurening forårsager kronisk stress hos fugle, med sundhedsmæssige konsekvenser for ungeDenne vestlige blåfugl vogter sin klø af æg i en redekasse i et naturgasfelt. Ude af stand til at skelne, om deres miljø er sikkert på grund af kompressorstøj, fuglemor skal vælge mellem at holde sig
- Er det nogle ting, vi genbruger bedre på lossepladser?
- Et stort scorekort giver det australske miljøs sundhed mindre end 1 ud af 10
- Nye elektronspin-hemmeligheder afsløret:Opdagelse af en ny sammenhæng mellem magnetisme og elektri…
- Kunder, der betaler for deres køb med kort, er mindre tilbøjelige til at huske det præcise betalt…
- Er mænd mere voldelige end kvinder?
- Muslimske LGBTQI+-flygtninge er mere tilbøjelige til at få asyl i Tyskland, hvis de overholder ste…