


Kunstig intelligens til at opnå kemiske fingeraftryk
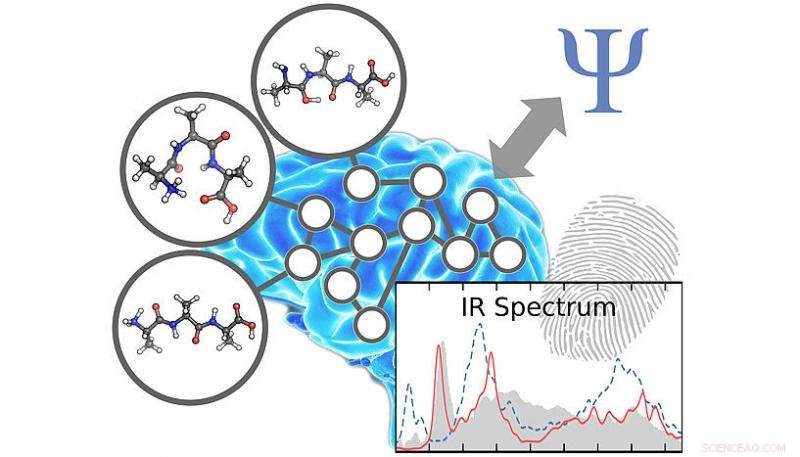
Forskerne har fundet en måde at fremskynde kemiske simuleringer ved hjælp af kunstig intelligens. Kredit:Philipp Marquetand
Det er lykkedes forskere ved universiteterne i Wien og Göttingen at udvikle en metode til at forudsige molekylære infrarøde spektre baseret på kunstig intelligens. Disse kemiske "fingeraftryk" kunne kun simuleres ved almindelige forudsigelsesteknikker for små molekyler i høj kvalitet. Ved hjælp af den nye teknologi, som er baseret på neuronale netværk svarende til den menneskelige hjerne og derfor er i stand til at lære, holdet ledet af Philipp Marquetand fra det kemiske fakultet ved universitetet i Wien var i stand til at udføre simuleringer, som tidligere ikke var mulige. Potentialet i denne nye strategi er nu blevet offentliggjort i det aktuelle nummer af tidsskriftet Kemisk Videnskab .
Drastiske fremskridt inden for forskning i kunstig intelligens har ført til en lang række fascinerende udviklinger på dette område i løbet af det sidste årti. Autonomt drevne biler, men også hverdagsapplikationer som søgemaskiner og spamfiltre illustrerer alsidigheden af metoder inden for kunstig intelligens.
Infrarød spektroskopi er en af de mest værdifulde eksperimentelle metoder til at få indsigt i molekylernes verden. Infrarøde spektre er kemiske fingeraftryk, der giver information om sammensætning og egenskaber af stoffer og materialer. I mange tilfælde, disse spektre er meget komplekse - en detaljeret analyse gør computerstøttede simuleringer uundværlige. Mens kvantekemiske beregninger i princippet muliggør ekstremt præcis forudsigelse af infrarøde spektre, deres anvendelighed i praksis vanskeliggøres af den høje beregningsmæssige indsats forbundet med dem. Af denne grund, pålidelige infrarøde spektre kan kun beregnes for relativt små kemiske systemer.
Forskerne har nu fundet en måde at accelerere disse simuleringer ved hjælp af kunstig intelligens. Til dette formål, såkaldte kunstige neurale netværk bruges, matematiske modeller af den menneskelige hjerne. Disse er i stand til at lære de komplekse kvantemekaniske forhold, der er nødvendige for modellering af infrarøde spektre, ved kun at bruge nogle få eksempler. På denne måde forskerne kan udføre simuleringer inden for få minutter, hvilket ellers ville tage tusinder af år selv med moderne supercomputere - uden at ofre pålideligheden. "Vi kan nu endelig simulere kemiske problemer, som ikke kunne overvindes med de hidtil brugte simuleringsteknikker, siger Michael Gastegger, undersøgelsens første forfatter.
Baseret på resultaterne af denne undersøgelse, forskerne er overbeviste om, at deres metode til spektreforudsigelse vil blive meget brugt i analysen af eksperimentelle infrarøde spektre i fremtiden.
 Varme artikler
Varme artikler
-
 Ny protonstarter til optogenetikKredit:MIPT Forskere har undersøgt et protein, der finder anvendelse i optogenetik og kan bruges til at kontrollere muskler og neuronale celler. Papiret om det lysfølsomme NsXeRprotein fra xenorho
Ny protonstarter til optogenetikKredit:MIPT Forskere har undersøgt et protein, der finder anvendelse i optogenetik og kan bruges til at kontrollere muskler og neuronale celler. Papiret om det lysfølsomme NsXeRprotein fra xenorho -
 Forskerhold finder mulig ny tilgang til sovesygemedicinStruktur af parasittens IMP-dehydrogenase. Det aktive enzym danner par (dimerer), switch-området (Bateman-regionen) vises i blå nuancer. Kredit:Lübeck Universitet/DESY, Lars Redecke Ved hjælp af u
Forskerhold finder mulig ny tilgang til sovesygemedicinStruktur af parasittens IMP-dehydrogenase. Det aktive enzym danner par (dimerer), switch-området (Bateman-regionen) vises i blå nuancer. Kredit:Lübeck Universitet/DESY, Lars Redecke Ved hjælp af u -
 Kombineret billeddannelsestilgang karakteriserer plaques forbundet med Alzheimers sygdomHistologi, FTIR, XFM, og vævsautofluorescensbilleddannelse af Aβ-plaques. Kredit:University of Adelaide Australske synkrotron røntgen og infrarød billeddannelsesteknikker er blevet brugt i en kraf
Kombineret billeddannelsestilgang karakteriserer plaques forbundet med Alzheimers sygdomHistologi, FTIR, XFM, og vævsautofluorescensbilleddannelse af Aβ-plaques. Kredit:University of Adelaide Australske synkrotron røntgen og infrarød billeddannelsesteknikker er blevet brugt i en kraf -
 Karamelreceptor identificeretDen lugtende furaneol giver jordbær og andre fødevarer en karamellignende aroma. Figur:G. Olias / LSB, Figuren viser jordbær og strukturformlen for lugtstoffet furaneol. Kredit:G. Olias / LSB Hvem
Karamelreceptor identificeretDen lugtende furaneol giver jordbær og andre fødevarer en karamellignende aroma. Figur:G. Olias / LSB, Figuren viser jordbær og strukturformlen for lugtstoffet furaneol. Kredit:G. Olias / LSB Hvem
- Kan fjernelse af kulstof fra atmosfæren redde os fra en klimakatastrofe?
- Opdagelse i nordlige søer kan være nøglen til at forstå det tidlige liv på Jorden
- Siliciumstrategi viser løfte om batterier
- Luftforureningsdata i fem kinesiske byer adskiller sig fra lokale VS amerikanske overvågningsstatio…
- Russisk raket vender tilbage til tjeneste med opsendelse af amerikansk satellit
- Børns fiktion om terror fører en ungdomstilbagemelding mod paranoia efter 11. september