


Forskere sekvenserer verdens største pangenom for at hjælpe med at låse op for genetiske mysterier bag finere silke

Fænotypisk mangfoldighed hos silkeorme. Kredit:BGI Genomics
BGI Genomics har i samarbejde med Southwest University, State Key Laboratory of Silkworm Genome Biology og andre partnere konstrueret et højopløseligt pangenom-datasæt, der repræsenterer næsten hele det genomiske indhold i en silkeorm.
Dette forskningspapir, der giver genetisk indsigt i kunstig selektion (domesticering og avl) og økologisk tilpasning, blev offentliggjort den 24. september i Nature Communications .
Tidligere, på grund af mangel på vilde silkeorme og tekniske begrænsninger i tidligere undersøgelser, manglede mange egenskabsrelaterede steder. Dette er den første forskning nogensinde til at digitalisere silkeorm-genpuljen og skabe en "digital silkeorm", hvilket i høj grad letter funktionel genomforskning, fremmer præcis avl og muliggør dermed yderligere silkebrug.
Holdet sekvenserer dybt 1.078 silkeorme (B. mori, inklusive 205 lokale stammer, 194 forbedrede sorter og 632 genetiske bestande og 47 vilde silkeorme, B. mandarina) og samler langlæste genomer på 545 af disse prøver, hvilket genererer 55,57 T-genomer. af genomiske data.
Dette pangenom-datasæt indeholder den mest omfattende information om genomerne af tamme og vilde silkeorm, og er det hidtil største langlæste pangenom i verden for planter og dyr. Samtidig er der udført dybdegående undersøgelser af forskellige genetiske variationer, befolkningsstruktur, kunstig selektion og økologiske tilpasninger og økonomiske træk ved silkeorm, hvilket har givet frugtbare resultater.
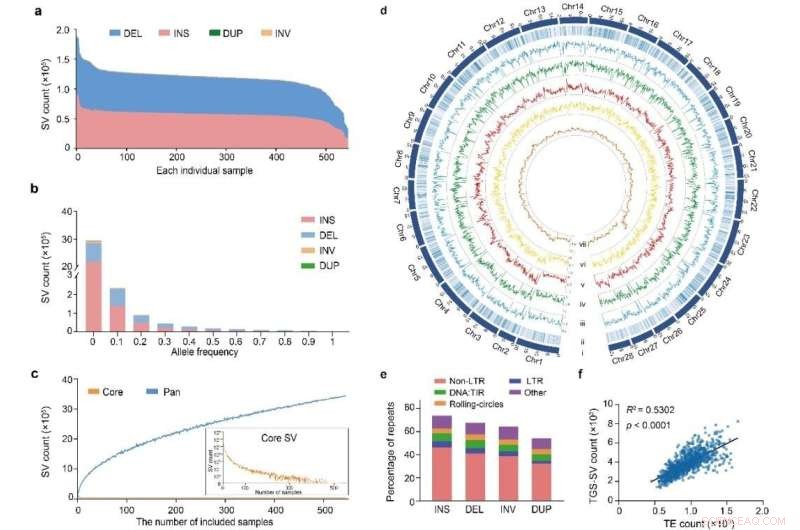
Karakterisering af strukturel variation i 545 silkeorms genomer. Kredit:BGI Genomics
Oprindelsen af den hjemlige silkeorm
Den tamme silkeorm, B. mori, tæmmet fra den vilde morbærsilkeorm, B. mandarina. Den har en historie på over 5.000 år, men dens hjemstedsplacering har længe været et åbent spørgsmål på grund af mangel på stærke biologiske beviser.
Materialet i denne undersøgelse repræsenterer den rigeste genetiske mangfoldighed fra alle større serikulturregioner i verden. Undersøgelsen fandt ud af, at endemiske arter fra Kinas nedre og midterste Yellow River-region er fordelt ved bunden af den indenlandske silkeormsgren på det evolutionære træ, hvilket tyder på, at den tamme silkeorm stammer fra denne region. De tilgængelige arkæologiske beviser, herunder en halv kokon udgravet i 1926 i Xiyin Village, Xia County, Shanxi-provinsen, og en stenudskåret silkeormspuppe udgravet i 2019 i Shicun i samme amt, giver vigtig støtte til denne konklusion.
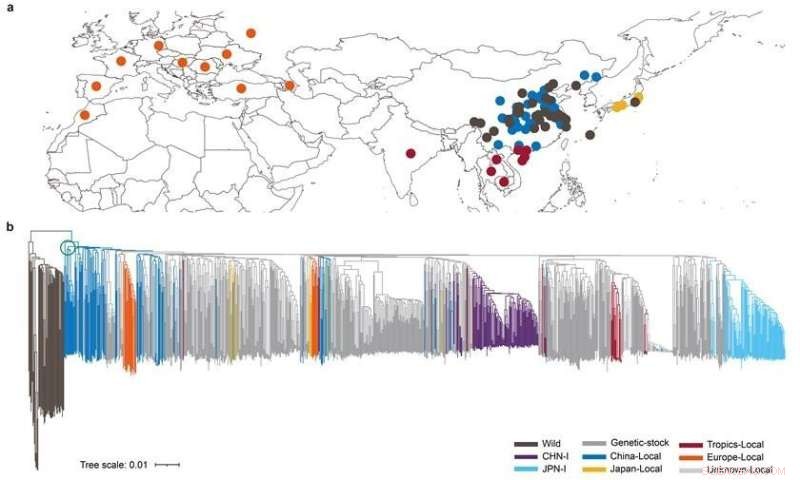
Geografisk fordeling og fylogenetisk træ af silkeorm. Kredit:BGI Genomics
At bryde flaskehalsen i silkeormeavl
Den traditionelle opdræt af silkeorme har en lang og unik historie, men siden 1990'erne forblevet fast i en flaskehals. Systematisk analyse af det genetiske grundlag for domesticering og forbedringsudvælgelse er afgørende for at løse de uløste problemer i silkeormeavl. Holdet har identificeret 468 domesticeringsassocierede gener og 198 forbedringsassocierede gener, hvoraf henholdsvis 264 og 185 er nyligt identificeret. Disse gener vil være vigtige kandidatmål for molekylær forbedring af silkeorm.
Samtidig blev det fundet ud af, at de kinesiske og japanske nyttearter deler mindre end 3 % af forbedringslocierne. Dette afslører ikke kun de relativt uafhængige avlshistorier for kinesisk og japansk silkeorm, men forklarer også, hvorfor dette fælles genetiske grundlag giver sådanne hybride fordele for begge arter. Dette resultat afgiver ny indsigt for fremtidig avl af silkeormen.
Økonomiske træk ved silkeormeavl
Udbytte og kvalitet af silke har længe været målrettet som de vigtigste økonomiske kriterier for kunstig udvælgelse af silkeorm. Indtil denne dato er der dog lidt kendt om, hvordan gener og loci styrer disse kvantitative egenskaber. Pangenomet er uden tvivl den 'nærmeste bro' mellem fænotyper, især komplekse træk.
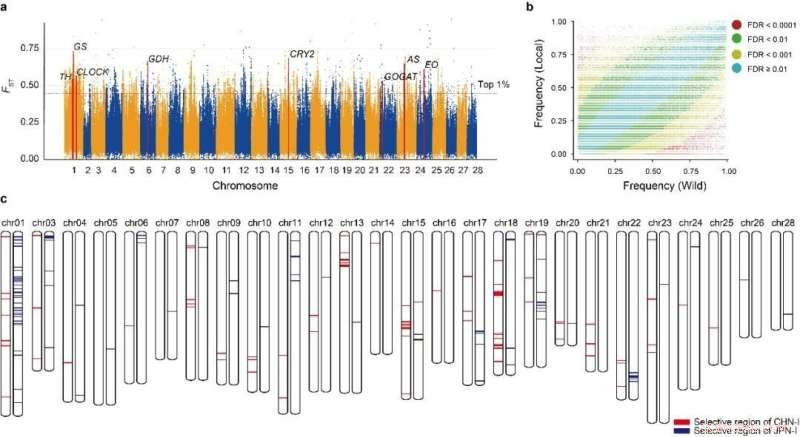
Silkeorms domesticering og avl. Kredit:BGI Genomics
Et eksempel på dette er reguleringen af silkeproduktion af den cellecyklus-relaterede transkriptionsfaktor BmE2F1, som blev afsløret gennem selektionssignalering og strukturel variation. CRISPR-cas9-medieret knockout af BmE2F1 reducerer antallet af silkekirtelceller med 7,68 % og silkeudbyttet med 22 %. Omvendt øger den transgene overekspression af BmE2F1 antallet af silkekirtelceller med 23 % og silkeudbyttet med 16 %.
Fin silke har unikke anvendelser og højere økonomisk værdi, men det genetiske grundlag for fiberfinhed forblev hidtil ukendt. Analyse af sjældne varianter i genomerne af slanke sorter førte til identifikation af BmChit β-GlcNAcase, et gen, der kontrollerer silkefinhed, der signifikant kan påvises i fine varianter, og CRISPR-cas9-medieret knockout, hvilket resulterer i grovere silkefinhed produceret af tamme silkeorme . Dette tyder på, at dette gen spiller en nøglerolle i bestemmelsen af silkefinhed.
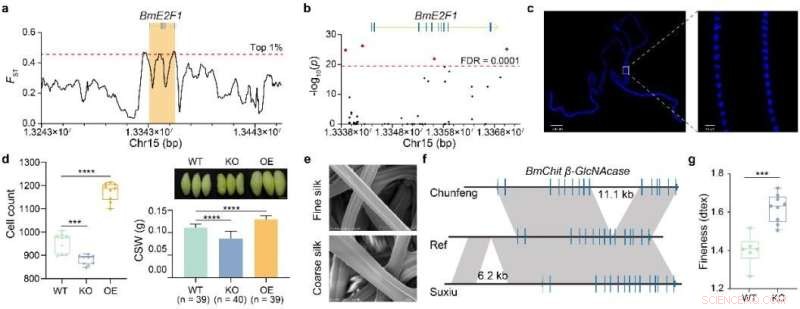
Genetisk grundlag for silkeorms økonomiske træk. Kredit:BGI Genomics
Tilpasning af silkeormeavl
Diapause er et almindeligt økologisk adaptivt træk hos insekter, der sikrer, at insekter kan overleve på trods af ugunstige miljøforhold. Selvom diapausehormonet først blev identificeret i silkeormen i 1957, er der kun få oplysninger om det embryonale dipause-gen. I denne undersøgelse, baseret på analysen af pnd-stammen og genomisk strukturel variation i silkeormen, og funktionel validering ved genredigering, viste det BmTret1-lignende gen sig at være en vigtig determinant for post-embryonal stalling. Det er første gang, at et post-embryonalt determinant gen er blevet identificeret i et insekt.
Denne undersøgelse afslører silkeormens komplette pan-genom for at låse op for kunstig selektion og økologisk tilpasningsindsigt. Shuaishuai Tai, medforfatter og BGI Genomics seniorforsker kommenterede:"Med omfattende prøveudtagning og datasæt kombineret med en række eksperimenter for at identificere gener til fremtidig potentiel undersøgelse, håber vi at fremskynde processen med silkeorms molekylært designopdræt." + Udforsk yderligere
Genetisk undersøgelse af silkeorm hjælper med at optrevle dens lange historie med domesticering
 Varme artikler
Varme artikler
-
 Hvor fermenteres fiber i grisenes fordøjelseskanal?Tamgris. Kredit:Scott Bauer, USDA Fiber tilsættes i stigende grad til svinefoder, men fordøjelsen af fibre i grise er ineffektiv og dårligt forstået. I en ny undersøgelse fra University of Illin
Hvor fermenteres fiber i grisenes fordøjelseskanal?Tamgris. Kredit:Scott Bauer, USDA Fiber tilsættes i stigende grad til svinefoder, men fordøjelsen af fibre i grise er ineffektiv og dårligt forstået. I en ny undersøgelse fra University of Illin -
 Ny undersøgelse beskriver, hvordan befrugtning udløser ændringer i tusinder af proteiner i frøæ…Overfladekontraktionsbølger på et frøæg udløses af befrugtning og drives af proteinaktivitet. Kredit:Tessa Montague I mere end et halvt århundrede, undersøgelser af den afrikanske klofrø (Xenopus
Ny undersøgelse beskriver, hvordan befrugtning udløser ændringer i tusinder af proteiner i frøæ…Overfladekontraktionsbølger på et frøæg udløses af befrugtning og drives af proteinaktivitet. Kredit:Tessa Montague I mere end et halvt århundrede, undersøgelser af den afrikanske klofrø (Xenopus -
 Blomster hemmeligt signal til bier og andre fantastiske nanoteknologier gemt i planterKredit:Shutterstock Blomster har et hemmeligt signal, der er specielt skræddersyet til bier, så de ved, hvor de skal samle nektar. Og ny forskning har netop givet os et større indblik i, hvordan d
Blomster hemmeligt signal til bier og andre fantastiske nanoteknologier gemt i planterKredit:Shutterstock Blomster har et hemmeligt signal, der er specielt skræddersyet til bier, så de ved, hvor de skal samle nektar. Og ny forskning har netop givet os et større indblik i, hvordan d -
 Hvad er de fem klasser af immunoglobuliner?Immunoglobuliner, også kaldet antistoffer, er glycoproteinmolekyler, der udgør en vigtig del af immunsystemet, der er ansvarlig for at bekæmpe infektionssygdom og udenlandske invasioner mere generelt.
Hvad er de fem klasser af immunoglobuliner?Immunoglobuliner, også kaldet antistoffer, er glycoproteinmolekyler, der udgør en vigtig del af immunsystemet, der er ansvarlig for at bekæmpe infektionssygdom og udenlandske invasioner mere generelt.
- Oversvømmelsesfrekvensen af verdens største flod er femdoblet
- Ulige-kronbladstilstande og vedvarende strømme i spin-kredsløbskoblede Bose-Einstein-kondensater
- Gedemælkskefir har vist sig at være godt for dit helbred
- I træg russisk økonomi, halal ser vækst
- Havrobotter tager pulsen på vores planet ved at måle mikrober
- Socialt samvær i COVID-tiden