


Beregning af fingeraftryk af molekyler med kunstig intelligens
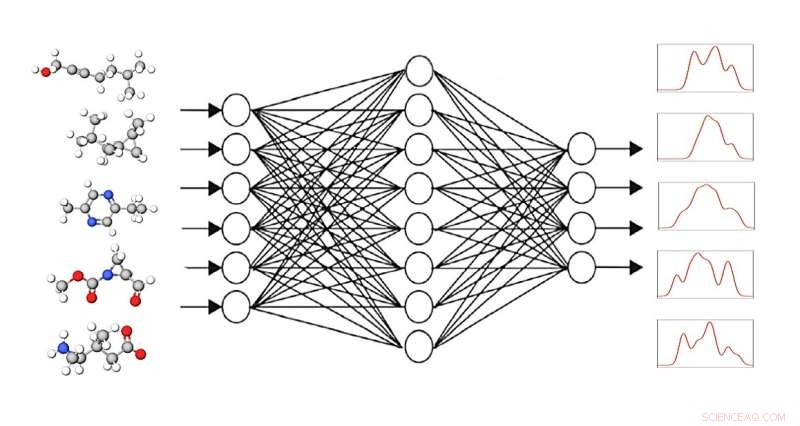
Det grafiske neurale netværk GNN modtager små molekyler som input med den opgave at bestemme deres spektrale responser. Ved at matche dem med de kendte spektre lærer GNN-programmet at beregne spektre pålideligt. Kredit:K. Singh, A. Bande/HZB
Med konventionelle metoder er det ekstremt tidskrævende at beregne det spektrale fingeraftryk af større molekyler. Men dette er en forudsætning for korrekt fortolkning af eksperimentelt opnåede data. Nu har et team hos HZB opnået meget gode resultater på væsentligt mindre tid ved at bruge selvlærende grafiske neurale netværk.
"Makromolekyler men også kvanteprikker, som ofte består af tusindvis af atomer, kan næppe beregnes på forhånd ved hjælp af konventionelle metoder som DFT," siger PD Dr. Annika Bande ved HZB. Sammen med sit team har hun nu undersøgt, hvordan regnetiden kan forkortes ved at bruge metoder fra kunstig intelligens.
Ideen:et computer "grafisk neuralt netværk" eller GNN modtager små molekyler som input med den opgave at bestemme deres spektrale responser. I næste trin sammenligner GNN-programmet de beregnede spektre med de kendte målspektre (DFT eller eksperimentelle) og korrigerer beregningsvejen i overensstemmelse hermed. Runde efter runde bliver resultatet bedre. GNN-programmet lærer således på egen hånd, hvordan man kan beregne spektre pålideligt ved hjælp af kendte spektre.
"Vi har trænet fem nyere GNN'er og fundet ud af, at der kan opnås enorme forbedringer med en af dem, SchNet-modellen:Nøjagtigheden øges med 20%, og dette gøres på en brøkdel af beregningstiden," siger førsteforfatter Kanishka Singh. Singh deltager i HEIBRiDS forskerskole og vejledes af to eksperter med forskellige baggrunde:datalogiekspert prof. Ulf Leser fra Humboldt University Berlin og teoretisk kemiker Annika Bande.
"Nyligt udviklede GNN-rammer kunne gøre det endnu bedre," siger hun. "Og efterspørgslen er meget stor. Vi ønsker derfor at styrke denne forskningslinje og planlægger at oprette en ny postdoktorstilling til den fra sommer og frem som en del af Helmholtz-projektet 'Forklarlig kunstig intelligens til røntgenabsorptionsspektroskopi'."
Forskningen blev offentliggjort i Journal of Chemical Theory and Computation . + Udforsk yderligere
Hvordan kvanteprikker kan 'tale' med hinanden
 Varme artikler
Varme artikler
-
 Ny fysiklov hjælper mennesker og robotter med at forstå friktionen ved berøringForskere har nu beskrevet en ny fysiklov, der forklarer elastohydrodynamisk smøring (EHL) friktion, som skulle fremme en bred vifte af robotteknologier. EHL-friktion opstår, når to faste overflader ko
Ny fysiklov hjælper mennesker og robotter med at forstå friktionen ved berøringForskere har nu beskrevet en ny fysiklov, der forklarer elastohydrodynamisk smøring (EHL) friktion, som skulle fremme en bred vifte af robotteknologier. EHL-friktion opstår, når to faste overflader ko -
 Gør almindelig mad ekstraordinærSidsteårs FST-studerende Chew Ding Xiang holder en flaske højtryksforarbejdet bok choyjuice. Denne forbedrede version bevarer næringsstoffer godt, og bevarer grøntsagernes smag. Kredit:National Univer
Gør almindelig mad ekstraordinærSidsteårs FST-studerende Chew Ding Xiang holder en flaske højtryksforarbejdet bok choyjuice. Denne forbedrede version bevarer næringsstoffer godt, og bevarer grøntsagernes smag. Kredit:National Univer -
 Analyse af fingermærker med synkrotronteknikker giver ny indsigtKredit:Australian Nuclear Science and Technology Organisation (ANSTO) Resultaterne af ledende forskere Prof Simon Lewis og Dr. Mark Hackett kan give muligheder for at optimere nuværende fingermærk
Analyse af fingermærker med synkrotronteknikker giver ny indsigtKredit:Australian Nuclear Science and Technology Organisation (ANSTO) Resultaterne af ledende forskere Prof Simon Lewis og Dr. Mark Hackett kan give muligheder for at optimere nuværende fingermærk -
 Kortlægning af elektriske felter for at hjælpe med at opklare, hvordan enzymer virkerGrafisk abstrakt. Kredit:Nature Chemistry (2022). DOI:10.1038/s41557-022-00937-w Hvert øjeblik i vores krops celler forekommer utallige aktiviteter, der er livsvigtige, takket være enzymer. Disse s
Kortlægning af elektriske felter for at hjælpe med at opklare, hvordan enzymer virkerGrafisk abstrakt. Kredit:Nature Chemistry (2022). DOI:10.1038/s41557-022-00937-w Hvert øjeblik i vores krops celler forekommer utallige aktiviteter, der er livsvigtige, takket være enzymer. Disse s
- Modellering tyder på at gå tidligt og gå hårdt vil redde liv og hjælpe økonomien
- NASA er ivrig efter at høre fra Mars rover, mens støvstormen forsvinder
- Hvordan sociale medier kan forbedre vores sproglige repertoirer
- Strain muliggør nye anvendelser af 2-D materialer
- Hvordan kometstøv afslører historien om solsystemet
- Forskere introducerer et nyt perspektiv inden for robotisk kapacitet